

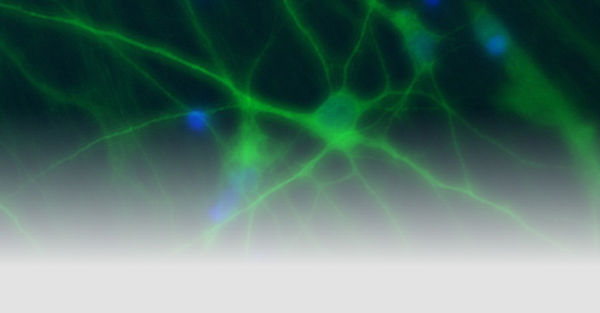
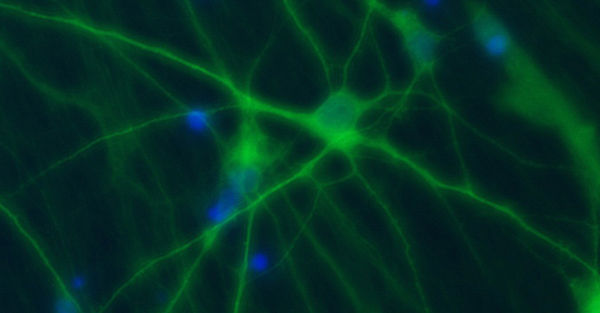
Ziel unserer Forschung ist es, die molekularen Mechanismen von hauptsächlich genetisch bedingten, neurologischen Erkrankungen, die durch eine abnormale neuronale Erregbarkeit verursacht werden, mit deren klinischen Symptomen und einer personalisierten Behandlung zu verknüpfen. Wir rekrutieren definierte Kohorten von Patienten mit Epilepsien und verwandten Erkrankungen, suchen mit modernen Sequenziermethoden nach krankheitsverursachenden Gendefekten, insbesondere in Ionenkanälen, Transportern und synaptischen Proteinen und analysieren deren funktionellen Konsequenzen, um die zugrunde liegenden Krankheitsmechanismen zu verstehen. Ein besonderer Schwerpunkt ist die Erforschung und Entwicklung neuer personalisierter Therapien für genetische Erkrankungen. Um die Mechanismen neuronaler Übererregbarkeit auf molekularer, zellulärer und Netzwerk-Ebene zu untersuchen, nutzen wir nicht-neuronale Modellsysteme wie automatisierte Messverfahren in Oozyten und Säugetierzellen, sowie neuronale Expressionssysteme. Letzteres beinhaltet die Generierung von Neuronen aus induzierten pluripotenten Stammzellen, die Verwendung menschlicher Hirnschnitte und genetischer Mausmodelle.
- Funktionelle Untersuchungen von genetischen Defekten in Ionenkanälen
Das BMBF geförderte Treat-ION-Konsortium für neurologische Ionenkanal- und Transporter-assoziierte Erkrankungen hat seinen Fokus auf therapeutischen Studien in zellulären, tierischen und humanen Modellsystemen, welche durch in-silico Experimente komplementiert werden. Diese zielen auf die Erforschung neuer Behandlungsmethoden sowie genauere Vorhersagen der funktionellen Folgen von Mutationen ab. Die Verwendung zugelassener und verfügbarer Medikamente (“repurposed drugs“) wie 4-Aminopyridin ist hierbei ein wesentliches Ziel, um eine präzise Behandlung zu ermöglichen. Durch die molekularen therapeutischen Gremien der Deutschen Akademie für seltene neurologische Erkrankungen (DASNE) und die Zentren für Seltene Erkrankungen (ZSE) in Baden-Württemberg werden unsere Ergebnisse direkt an die Patienten übermittelt. Die funktionellen Auswirkungen ausgewählter Mutationen werden in neuronalen Expressionssystemen, wie transfizierten Primärneuronen der Maus, in-utero elektroporierten Neuronen und genetisch veränderten Tiermodellen, die eine humane Mutation tragen (sogenannte "humanisierte Mausmodelle"), untersucht. Der Vorteil sowohl von in-utero elektroporierten Neuronen als auch von genetisch veränderten Mausmodellen ist, dass Ionenkanäle mit einer pathologischen Variante unter physiologisch relevanten Bedingungen untersucht werden. Mittels Patch-Clamp Messungen können hierbei die intrinsischen, neuronalen Eigenschaften einzelner Neurone untersucht werden, wohingegen extrazelluläre Ableitungen sowie Multielektrodenarrays (MEA) zur Erforschung der Netzwerkaktivität dienen. Die Verwendung von 256-Elektroden MEAs sowie hochauflösender CMOS-Chips mit 4000 Elektroden erlaubt es uns einzelne Zellkompartimente und neuronale Netzwerkaktivität in Hirnschnitte von transgenen Tieren zu analysieren. Im Rahmen der DFG-Forschergruppe und insbesondere in Zusammenarbeit mit Prof. Dr. Garaschuk nutzen wir zudem in-vivo Ca2+-Imaging, um die Netzwerkdysfunktionen unserer Mausmodelle weiter zu erforschen. Um genauer zu verstehen, wie in Folge eines genetischen Defekts Epilepsie entsteht, untersuchen wir die regionale und zeitspezifische RNA Expression mittels Einzelzell-RNA-Sequenzierung in verschiedenen neuronalen Subpopulationen der Mausmodelle.
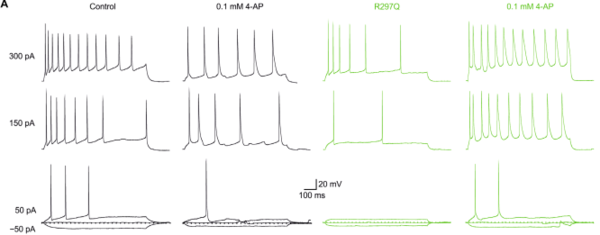
Patch-Clamp Messungen von Mausneuronen vor und nach der Zugabe von 100 µM 4-AP. Schwarz: Neuron, welches einen KV1.2-Wildtyp (durch KCNA2 codiert) exprimiert. Grün: Neuron, welches eine KV1.2-Gain-of-function Variante exprimiert. - Induzierte pluripotente Stammzellen als Epilepsiemodelle
Da humanes Hirngewebe eine limitierte Ressource in der neurowissenschaftlichen Forschung ist, generieren wir zusätzlich Neurone aus induzierten pluripotenten Stammzellen (iPSC). Diese stellen eine attraktive, wenn auch vereinfachte, Alternative eines humanen Modellsystems zur Untersuchung von Erkrankungen des Nervensystems dar.
Durch die Reprogrammierung patientenspezifischer Fibroblasten in pluripotente Stammzellen und die darauffolgende Überexpression spezifischer Transkriptionsfaktoren können wir gezielt exzitatorische und inhibitorische kortikale Neurone generieren. In anschließenden funktionellen Studien kann der Krankheitsmechanismus einzelner Patienten unter Berücksichtigung ihres einzigartigen genetischen Hintergrunds untersucht werden. Dabei untersuchen wir sowohl die Netzwerkaktivität mittels eines Multiwell-MEA-Systems, als auch die Einzelzellaktivität mittels Patch-Clamp Messungen. Somit konnten wir bereits zeigen, dass die entwicklungsbedingten elektrophysiologischen Aktivitätsmuster in iPSC-Neuronen mit denen von Menschen und Tieren vergleichbar sind (Rosa et al., Stem Cell Rep 2020). Aktuelle Projekte umfassen die elektrophysiologische Analyse patientenspezifischer iPSC-Neurone, welche pathologische Mutationen in Genen für Ionenkanäle (KCNA2, KCNH1) oder synaptische Proteine (STX1B) tragen.
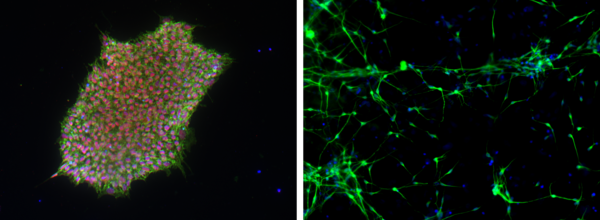
Expression der Marker SOX2 (grün) und SEEA4 (rot) weist Pluripotenz der iPSCs nach (links). Neurone (grün) aus induzierten pluripotenten Stammzellen, zwei Wochen nach spontaner Embryoid body Differenzierung (rechts). - Humane Hirnschnittkulturen
Als weiteres humanes Modellsystem verwenden wir humane Schnittkulturen. Bei der Verwendung von humanem Liquor (CSF) als Kulturmedium, können die Schnittkulturen bis zu vier Wochen mit guten neurophysiologischen Eigenschaften in Kultur gehalten werden. Die Verwendung von ex-vivo-Gehirnschnitten, die aus adultem humanem neurochirurgisch reseziertem Gewebe stammen, ermöglicht die Untersuchung elektrophysiologischer Eigenschaften auf Einzelzell- und Netzwerkebene (Schwarz et al., Sci Rep 2017). Mit dieser Methode konnten wir erstmals zeigen, dass sowohl die neuronale Zytoarchitektur als auch die elektrophysiologischen Eigenschaften menschlicher Pyramidenzellen, wie auch Interneurone, stabil über 14 Tage erhalten bleiben. In weiteren Experimenten konnte durch eine optimierte virale Transduktion der Schnittkulturen ein High-throughput Verfahren für 3D-Rekonstruktionen GFP-markierter Zellen etabliert werden welche zukünftige Langzeit live Imaging von menschlichen Zellen in-vitro ermöglicht.

Nach neurochirurgischen Eingriffen wird Hirngewebe, welches für einen nötigen Zugang entfernt werden muss, in kleine Stücke geschnitten (links) und über Wochen kultiviert. Die Neurone behalten dabei ihre besondere Morphologie (rechts).

PhD S
tudent
+49 (0)7071-
29-81914

+49 (0) 7071
29-80442
+49(0)7071-
29-81914
+49 (0)7071-
29-81914

+49 (0)7071-
29-81914
+49 (0) 7071-
29-70868
+49 (0) 7071
29-87639
+49 (0) 7071
2-981983
+49 (0)7071
29 81914

+49 (0) 7071
29-81914

+49(0)7071-
29-85247

+49 (0) 7071
29-87638

+49 (0)7071-
29-81921
+49 (0)7071
29-81983

+49 (0) 7071
29-81984
+49 (0)7071-
29-81921
+49 (0) 7071
29-86525

+49 (0) 7071
2981914
+49 (0)7071-
29-87638

+49 (0) 7071
29-86588
+49 (0) 7071
29-80442
+49 (0)7071-
29-81921

+49 (0)7071-
29-81983

+49 (0) 7071
29-80442

+49 (0) 7071
29-81921
+49 (0) 7071
29-81914

+49 (0) 7071
29-81922
+49 (0) 7071
29-80440

+49 (0) 7071
29-80440
+49 (0) 7071
29- 81968
+49 (0)7071
29-81914
+49 (0) 7071
- 29- 70868

+49(0)7071
29- 80442

+49 (0) 7071-
29-81914
+49 (0)7071
29-81921
+49 (0) 7071-
29 81914
+49 (0)7071-
29-80440
'+49 (0)7071-
29-87639
+49 (0) 7071
29-81914
Ausgewählte Publikationen
Müller P*, Takacs DS*, Hedrich UBS, Coorg R, Masters L, Glinton KE, Dai H, Cokley JA, Riviello JJ, Lerche H#, Cooper EC#. KCNA1 gain-of-function epileptic encephalopathy treated with 4-aminopyridine. Ann Clin Transl Neurol. 2023 Feb 15.
Krüger J, Schubert J, Kegele J, Labalme A, Mao M, Heighway J, Seebohm G, Yan P, Koko M, Aslan-Kara K, Caglayan H, Steinhoff BJ, Weber YG, Keo-Kosal P, Berkovic SF, Hildebrand MS, Petrou S, Krause R, May P, Lesca G, Maljevic S, Lerche H. Loss-of-function variants in the KCNQ5 gene are implicated in genetic generalized epilepsies. EBioMedicine. 2022 Oct;84:104244. doi: 10.1016/j.ebiom.2022.104244.
Boßelmann CM, Hedrich UBS, Müller P, Sonnenberg L, Parthasarathy S, Helbig I, Lerche H, Pfeifer N. Predicting the functional effects of voltage-gated potassium channel missense variants with multi-task learning. EBioMedicine. 2022 Jul;81:104115.
Johannesen KM*, Liu Y*, Koko M, Gjerulfsen CE, Sonnenberg L, Schubert J, Fenger CD, Eltokhi A, Rannap M, Koch NA,Lauxmann S, Krüger J, Kegele J, (…),Hedrich UBS, Benda J, Gardella E,Lerche H#, Møller RS#. Genotype-phenotype correlations in SCN8A-related disorders reveal prognostic and therapeutic implications. Brain 2021:awab321.
Auffenberg E*, Hedrich UB*, Barbieri R*, Miely D*, Groschup B, Wuttke TV, (…), Pusch M, Dichgans M, Lerche H, Gavazzo P#, Plesnila N#, Freilinger T#. Hyperexcitable interneurons trigger cortical spreading depression in an Scn1a migraine model. J Clin Invest 2021;131:e142202.
Hedrich UBS*, Lauxmann S*, (…), Bosselmann C, Schwarz N, Fudali M, Lerche H. 4-Aminopyridine is a promising treatment option for patients with gain-of-function KCNA2-encephalopathy. Sci Transl Med 2021;13:eaaz4957.
Koko M, Krause R, Sander T, Bobbili DR, Nothnagel M, May P,Lerche H; Epi25 Collaborative. Distinct gene-set burden patterns underlie common generalized and focal epilepsies. EBioMedicine 2021;72:103588.
Rosa F, Dhingra A, Uysal B, Mendis GDC, Loeffler H, Elsen G, Mueller S, Schwarz N, Castillo-Lizardo M, Cuddy C, Becker F, Heutink P, Reid CA, Petrou S, Lerche H#, Maljevic S#. In Vitro Differentiated Human Stem Cell-Derived Neurons Reproduce Synaptic Synchronicity Arising during Neurodevelopment. Stem Cell Reports 2020;15:22-37.
Schwarz N, Hedrich UBS, Schwarz H, P A H, Dammeier N, Auffenberg E, Bedogni F, Honegger JB, Lerche H, Wuttke TV, Koch H. Human Cerebrospinal fluid promotes long-term neuronal viability and network function in human neocortical organotypic brain slice cultures. Sci Rep. 2017 Sep 25;7(1):12249.
Coming soon

Hertie-Zentrum für Neurologie
Hertie-Institut für klinische Hirnforschung
Abteilung Neurologie mit Schwerpunkt Epileptologie
Hoppe-Seyler-Straße 3
72076 Tübingen
Tel.: +49 (0)7071 29-80466
Fax: +49 (0)7071 29-4488
Sabrina Kreiser
Yvonne Brändle
Tel: +49 (0)7071 29-80442
Fax: +49 (0)7071 29-4488
sekretariatne5.HL(at)med.uni-tuebingen.de
Heidrun Löffler
Tel: +49 (0)7071 29-81922
heidi.loeffler(at)medizin.uni-tuebingen.de




