


Neurogenetic diseases like cerebellar ataxia, hereditary spastic paraplegia and leukodystrophy are rare disorders. For many of them the causative gene is still not known. We aim to discover the genetic cause as this offers not only a definite diagnosis for our patients but also opens a window into pathogenesis and potential interventions in early stages of the disease process. To this end we are modelling neurogenetic diseases using induced pluripotent stem cells and its differentiation into neurons which are genetically identical to our patients.
In 2010 we founded the first Center for Rare Diseases (ZSE) in Germany with support from the former first lady Eva Luise Köhler. The ZSE is dedicated to translational research. Many patients with rare movement disorders are seen in the specialized outpatient clinics. Clinical research builds up representative cohorts ready for trials with comprehensive phenotyping and longitudinal data on the natural course of disease as well as biobanking for biomarker development.
We provide specialized outpatient clinics for diagnostic and therapy of rare movement disorders at the Department of Neurology:
- Center for rare neurological diseases (Huntington disease, mitochondriopathies, etc.)
Our clinical research aims to prepare the way towards new therapies. For this purpose several steps are required:
- Representative cohorts are a basic requirement for the development of new therapiesespecially in rare diseases. Funded by the German-HSP-patient-support group we establish a web-based registry for hereditary spastic paraplegia including a standardised recording of clinical and genetic data. Data of the baseline analysis is now available (Schüle et al., 2016). Follow-up examinations will provide prospective data about the progress of the disease in its natural course. Such data is essential in the planning of interventional trials which aim to slow down the course of the disease. Similar studies are carried out for Friedreich’s ataxia and other early onset ataxias, spinocerebellar ataxias, multiple system atrophy as well as adult forms of leukodystrophy.

- As clinical progression is rather slow and depends on many external factors, biomarkers that reflect disease activity are urgently needed. For hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) we are assessing cytokine profiles in blood and CSF reflecting abnormal processes in HDLS pathogenesis. Similarly, biomarkers are also developed to measure progression in ataxias and HSP.

- Ideally, therapeutical approaches should start before the onset of symptoms and postpone or prevent disease manifestation. In autosomal dominantly inherited diseases like HDLS, SPG4 and spinocerebellar ataxias (SCA) first degree relatives of patients have a 50% risk to carry the disease causing mutation. In depth examinations of such individuals at risk can help to identify markers that indicate disease activity in presymptomatic stages of the disease before the patients experience any impairment. We set up studies to define presymptomatic biomarkers in healthy mutation carriers of HDLS as well as SPG4 (preSPG4 study) and spinocerebellar ataxias (RISCA study; Jacobi et al., 2015) by combining computerized movement recording, elaborated MRI techniques and analyses of disease proteins (Lindig et al., 2015).

- The major goal of this research to establish trial ready cohorts that are suitable for interventional trials with promising compound identified in experimental approachestypo3/#_msocom_1 performed in parallel in our lab. Following this line, we recently performed the first randomized placebo-controlled trial in hereditary spastic paraplegia type 5 (SPG5) (Schöls et al., 2017).

The Schöls lab takes advantage of the broad access to patient-derived biomaterials and focusses on the identification of new disease genes and disease modelling by using induced pluripotent stem cells (iPSCs).
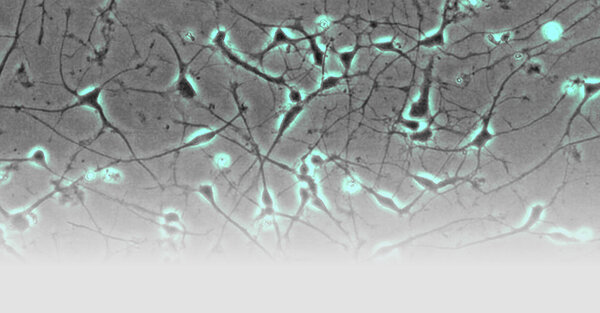
- Fibroblasts are reprogrammed into induced pluripotent stem cells (iPSC) by nucleofection of episomal plasmids carrying classic reprogramming factors (Fig. 1; Hauser et al., 2016). These stem cells can be re-differentiated into neurons or other tissue relevant for pathogenesis (e.g. hepatocytes). This approach allows to analyze neurons or other cell types in the culture dish that are genetically identical to our patients.

- iPSC-derived neurons are used to model the respective disease. E.g. hereditary spastic paraplegia preferentially affects the long axons of the cortico-spinal tract. Using live cell imaging and high content imaging we could demonstrate impaired axonal outgrowth in SPG4 neurons, the most common form of hereditary spastic paraplegia (in cooperation with Prof. Dr. Oliver Brüstle, Bonn). Furthermore, SPG4 axons present with largely increased numbers of axonal swellings compared to control neurons.

- SPG5 is a subtype of hereditary spastic paraplegia caused by mutations in the cytochrome CYP7B1. Lack of CYP7B1 in the liver leads to metabolic abnormalities with extensive increase of oxysterols in blood but also in CSF. Treating iPSC-derived neurons with oxysterols in concentrations that are found in our patients impairs axonal growth as well as cell viability. Currently we are characterizing metabolic abnormalities in iPSC-derived hepatocytes of SPG5 patients using mass spectrometry in cooperation with Prof. Ingemar Björkhem (Karolinska Institute, Stockholm) and Prof. William Griffiths (Swansea, UK).
- Genetic studies aim to disclose the genetic cause in patients with ultra-rare diseases. With funding from the DFG we run a trilateral project connecting Israeli, Palestinian and German scientists to discover new genes in so far undefined disorders . Genetic diseases are frequent in Israel and the Palestinian authorities due to numerous consanguineous marriages. Whole exome sequencing allowed us to identify several new genes like AP4B1, WWOX and DNAJC3 (Bauer et al. 2012; Mallaret et al. 2014; Synofzik et al. 2014). Genomic approaches are performed in collaborative European projects like NEUROMICS in close cooperation with Prof. Peter Bauer and Prof. Olaf Rieß (Institute of Medical Genetics, Tübingen), Prof. Michel Koenig (Montpellier), Dr. Holger Prokisch (Helmholtz Centre, Munich) and Prof. Stephan Züchner (Hussman Institute, Miami).


+49 (0)7071-
29-82057

+49 (0)7071-
9254-054

+49 (0)7071-
29-85653

+49(0)7071-
29-85170

+49 (0)7071-
29-87654

+49(0)7071-
29-85247

+49(0)7071-
9254-402

+49(0)7071-
29-85247

+49 (0)7071-
29-81969

+49(0)7071-
9254-054

+49(0)7071-
29-87609

+49 (0)7071-
29-85347
+49 (0)7071-
29-82057

+49 (0)7071-
29-85347

+49(0)7071-
29-87609

+49(0)7071-
29-87609

+49(0)7071-
29-82057
+49 (0)7071-
29-85374

+49(0)7071-
29-82057

+49 (0)7071-
29-85247

+49(0)7071-
9254-054

+49 (0)7071-
29-85247

+49(0)7071-
29-85247
Selected publications
Hengel H, Hannan SB, Dyack S, …, Schuster S, Hauser S, Admard J, Casadei N, Velic A, Macek B, Ossowski S, Houlden H, Maroofian R, Schöls L. Bi-allelic loss-of-function variants in BCAS3 cause a syndromic neurodevelopmental disorder. Am J Hum Genet. 2021;108:1069-1082
Collier JJ, Guissart C, Oláhová M, …, Schöls L, … Taylor RW. Developmental consequences of defective ATG7-mediated autophagy in humans. N Engl J Med. 2021; 384:2406-2417
Hengel H, Bosso-Lefèvre C, Grady G, … , Schöls L#, Reversade B#. Loss-of-function mutation in UDP-Glucose-6-Dehydrogenase cause recessive developmental epileptic encephalopathy. Nat Comm. 2020;11:595
Hauser S, Schuster S, Heuten E, Hoeflinger P, Admard J, Schelling Y, Velic A, Macek B, Ossowski S, Schöls L. Comparative Transcriptional Profiling of Motor Neuron Disorder-Associated Genes in Various Human Cell Culture Models. Frontiers Cell Dev Biol. 2020; ;8:544043
Synofzik M, Puccio H, Mochel F, Schöls L. Autosomal recessive cerebellar ataxias: paving the way towards targeted molecular therapies. Neuron. 2019;101:560-83
Hauser S, Poenisch M, Schelling Y, Hoflinger P, Schuster S, Teegler A, Betten R, Gustafsson JA, Hubener-Schmid J, Schlake T, Chevessier-Tunnesen F, Horscroft N, Bjorkhem I, Schöls L. mRNA as a Novel Treatment Strategy for Hereditary Spastic Paraplegia Type 5. Mol Ther Methods Clin Dev. 2019;15:359-370
Diallo A, Jacobi H, Cook A, Labrum R, Durr A, Brice A, Charles P, Marelli C, Mariotti C, Nanetti L, Panzeri M, Rakowicz M, Sobanska A, Sulek A, Schmitz-Hubsch T, Schöls L, Hengel H, Melegh B, Filla A, Antenora A, Infante J, Berciano J, van de Warrenburg BP, Timmann D, Boesch S, Pandolfo M, Schulz JB, Bauer P, Giunti P, Kang JS, Klockgether T, du Montcel ST. Survival in patients with spinocerebellar ataxia types 1, 2, 3, and 6 (EUROSCA): a longitudinal cohort study. Lancet Neurol. 2018;17:327-34
Schöls L, Rattay TW, Martus P, Meisner C, Baets J, Fischer I, Jagle C, Fraidakis MJ, Martinuzzi A, Saute JA, Scarlato M, Antenora A, Stendel C, Hoflinger P, Lourenco CM, Abreu L, Smets K, Paucar M, Deconinck T, Bis DM, Wiethoff S, Bauer P, Arnoldi A, Marques W, Jardim LB, Hauser S, Criscuolo C, Filla A, Zuchner S, Bassi MT, Klopstock T, De Jonghe P, Bjorkhem I, Schule R. Hereditary spastic paraplegia type 5: natural history, biomarkers and a randomized controlled trial. Brain. 2017;140:3112-7
Schüle R, Wiethoff S, Martus P, Karle KN, Otto S, Klebe S, Klimpe S, Gallenmüller C, Kurzwelly D, Henkel D, Rimmele F, Stolze H, Kohl Z, Kassubek J, Klockgether T, Vielhaber S, Kamm C, Klopstock T, Bauer P, Züchner S, Liepelt-Scarfone I, Schöls L. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann Neurol. 2016;79:646-58
Schöls L, Reimold M, Seidel K, Globas C, Brockmann K, Hauser TK, Auburger G, Bürk K, den Dunnen W, Reischl G, Korf HW, Brunt ER Rüb U. No parkinsonism in SCA2 and SCA3 despite severe neurodegeneration of the dopaminergic substantia nigra. Brain. 2015;138:3316-26





